1. Welcome
This is the MetaDraft user guide. MetaDraft is part of the CBMPy MetaToolkit project and is developed by the Systems Bioinformatics group at the VU University Amsterdam. For more information on MetaDraft please visit the MetaDraft pages at https://systemsbioinformatics.github.io/cbmpy-metadraft/#. MetaDraft is primarily written in Python and uses Qt for its GUI, as well as CBMPy for its object model and SBML support.
For information on installing MetaDraft and its dependencies please see the readme.md file included in your download or on GitHub, you will also find information on the systemtest.py utility that will check whether your system us ready to run MetaDraft.
2. Main reconstruction screen
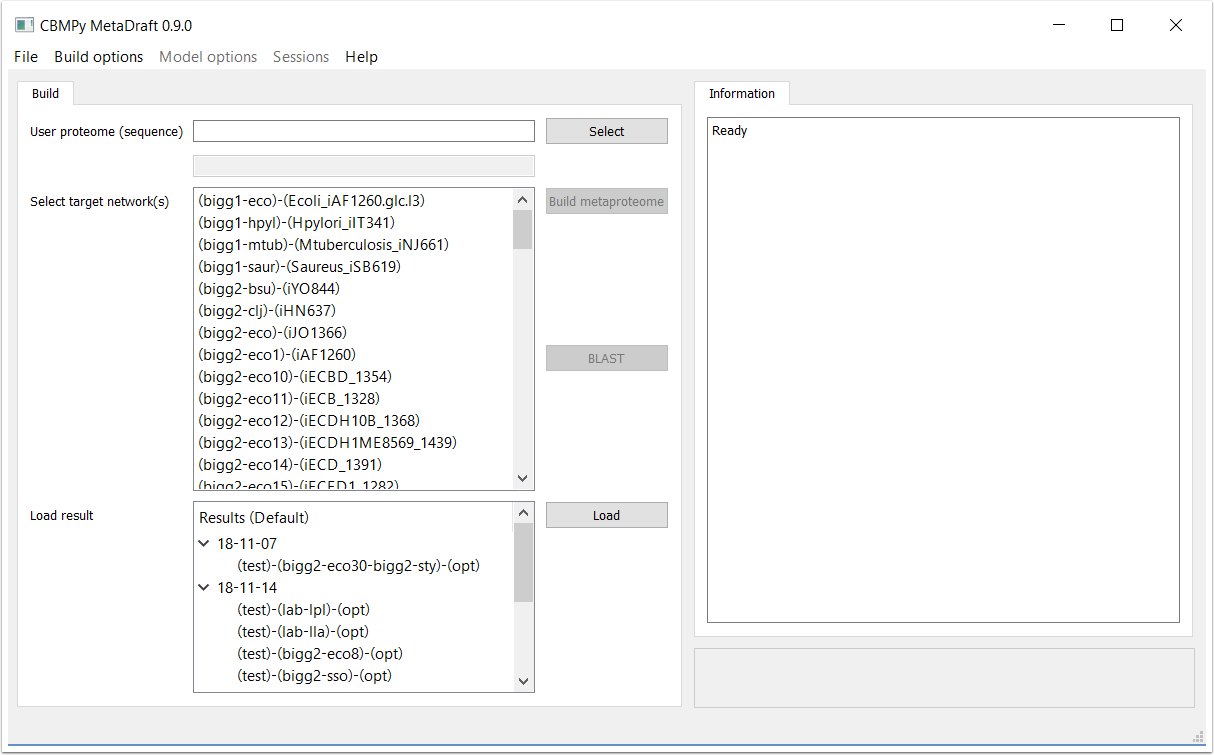
When MetaDraft starts up you will find yourself at the reconstruction screen. Here you are able to input the protein sequence file (either a protein FASTA or GenBank format file with CDS annotations), create a template database that you will BLAST your sequence against. Using trhe "Build Option" menu you can create your own, user-defined, template models and create custom sets of template models. You can also reload previously generated draft models in the "Load result" section.
2.1 Load your sequences
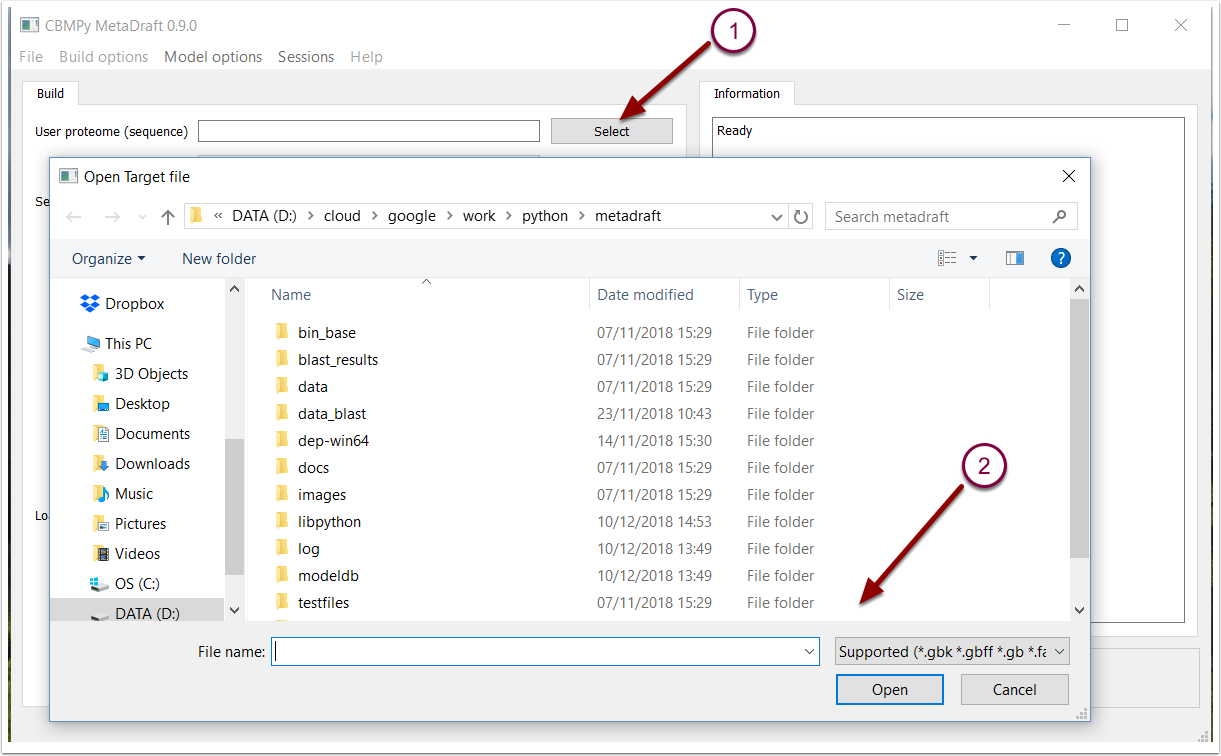
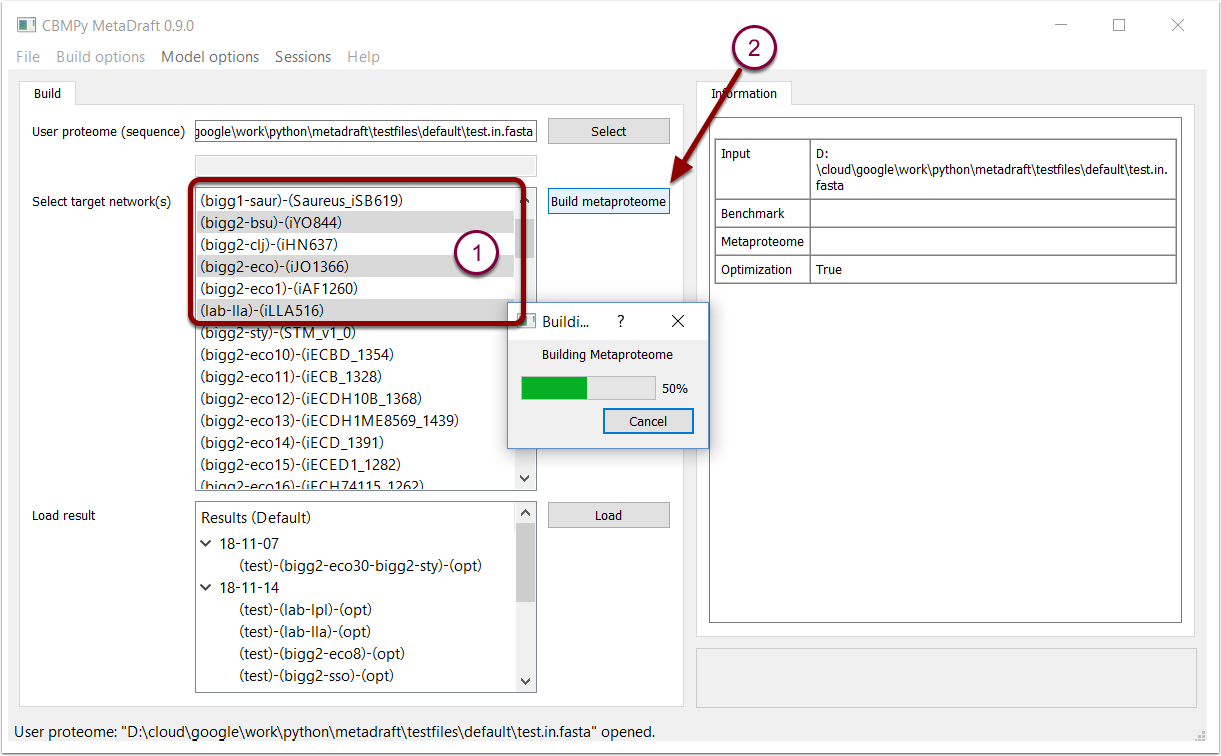
Let's start by loading the sequence file that you would like to turn into a draft model reconstruction. MetaDraft is able to process protein FASTA (*.fasta) or GenBank format files (that include CDS annotation), to load your file mouse over to the "User proteome (sequence)" field and hit the associated "Select button" (1). This will trigger a FileOpen window where you may select the appropriate, supported file (2).
2.2 Build a metaproteome from template models
MetaDraft works by comparing your input sequence to a database of known models and sequences or "template models". However, the modeller has control over the number and priority, of the template models that are assembled into what MetaDraft calls a metaproteome. In this release template models are created from the BiGG database and thus use a consistent set of reaction and metabolite identifiers, this is not a prerequisite for user-defined templates.
Once a user sequence has been input (previous step) one or more template models in the "Target network" list can be selected to form the basis of the metaproteome (1). Note that order does matter, template models higher in the list have priority when "ID optimization" is selcted. Template model priority can include factors such as, phylogenetic distance from your source model, phsyiological factors or template models quality/completeness.
Once a selection has been made the metaproteome can be constructed by pressing the "Build metaproteome" button (2).
2.3 Template model options
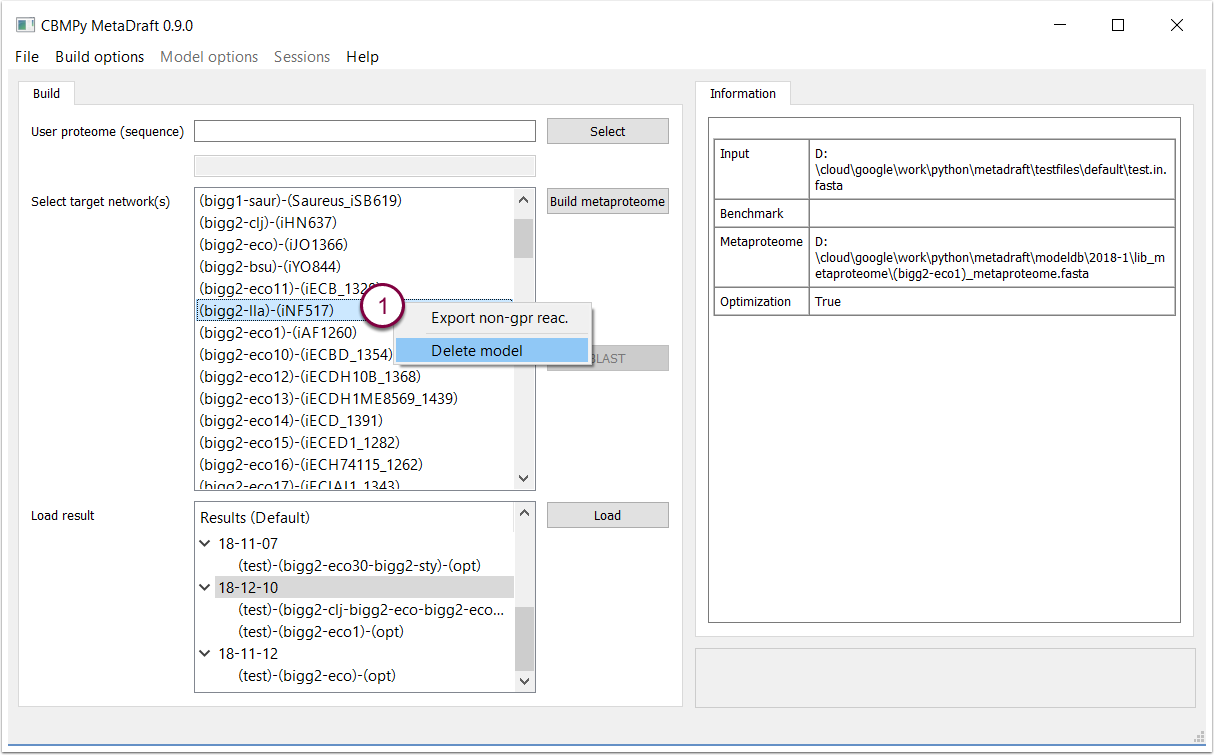
Right-clicking on a template model allows you to delete it from the model list or export the model reactions that are not associated with genes via a GPR association (1). Non-GPR reactions will be exported as an SBML Level 3 FBC file that can be used later in the reconstruction process.
2.4 User defined metaproteome selections
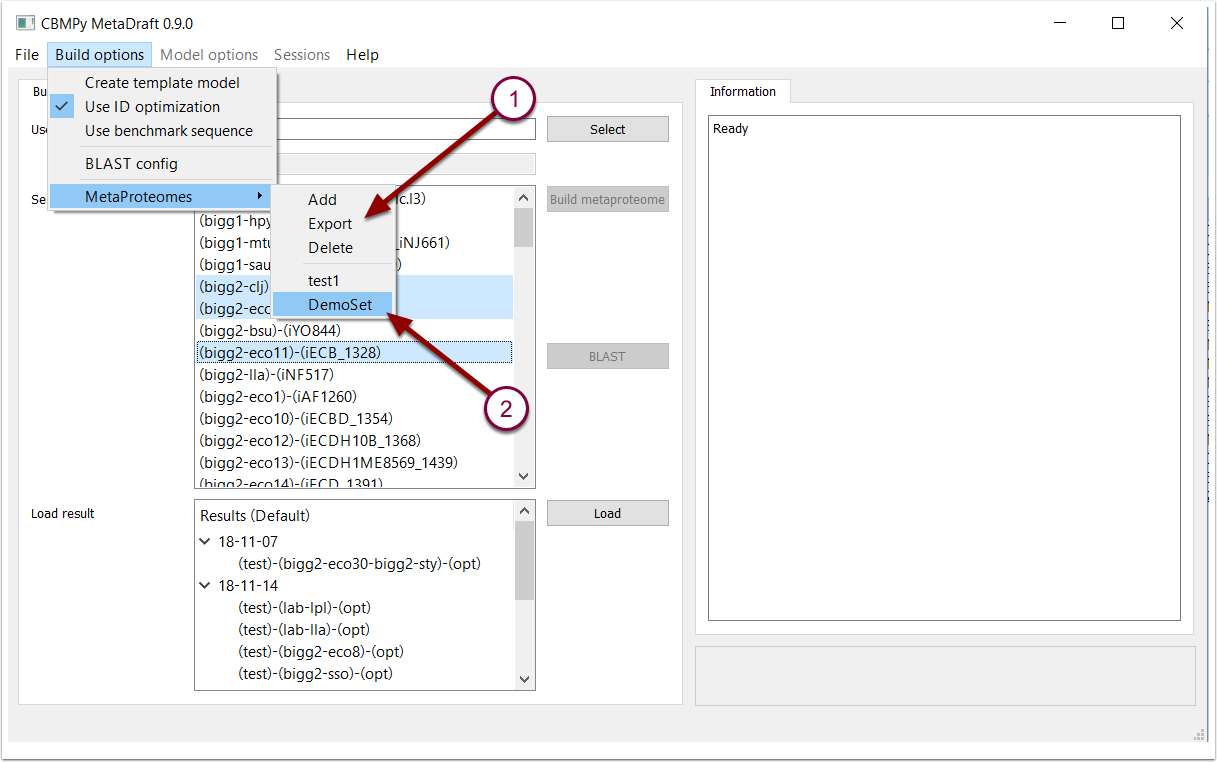
MetaDraft allows users to define their own metaproteomes which, once defined, can easily be selected from the "Build options" menu. Selecting the "MetaProteomes" sub-menu (1) you can "Add" or "Delete" metaproteomes or "Export" a metaproteome in FASTA format. This allows metaproteomes to be exported and used for other purposes outside of MetaDraft. Once defined metaproteomes become available for selection in the lower half of the "Metaproteomes" menu (2).
2.5 Metaproteome optimization
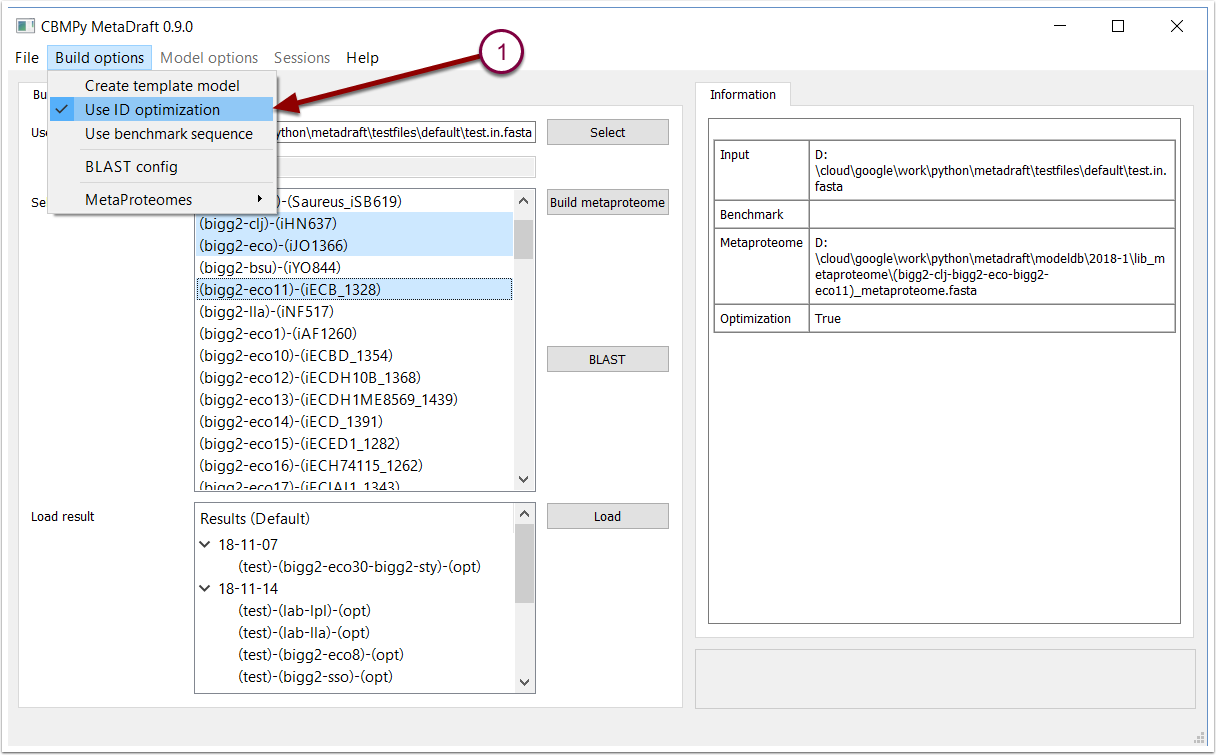
By default MetaDraft uses ID optimization to reduce the number of related reactions present in the metaproteome. ID optimization works best when all the template models in the metaproteome use a common reaction and (less important) metabolite set. In this optimization the highest ranking model (top selection in the template model list) is used as a starting point, for any addtional template only the reactions which are not present in the base template are added to the metaproteome.This option may easily be toggled on/off by selecting the "Use ID optimization" item in the "Build options" menu (1).
2.6 Run sequence search
Once a user proteome has been loaded and a metaproteome defined the "BLAST" button becomes active (1). Pressing this will initiate the orthology mapping, a process which might take minutes or hourse depending on the size of the user proteome and metaproteomes. On certain operating systems (some flavours of Linux)
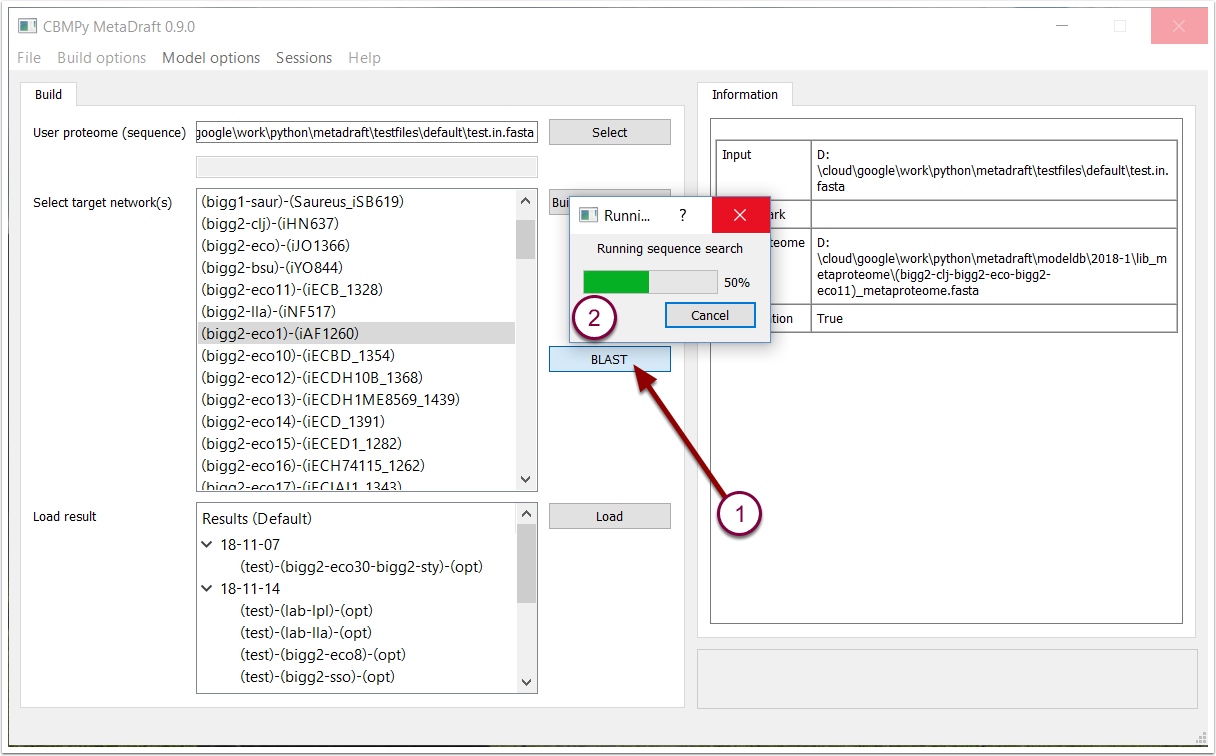
3. BLAST search results
When a BLAST search is complete or when MetaDraft is started up, the results of all previously run BLAST searches are shown in the results panel (1). By default these results are grouped by creation data with the results listed as (user proteome)-(metaproteome)-(optimization). To load a results, select the results (2) and press the "Load button" (3).
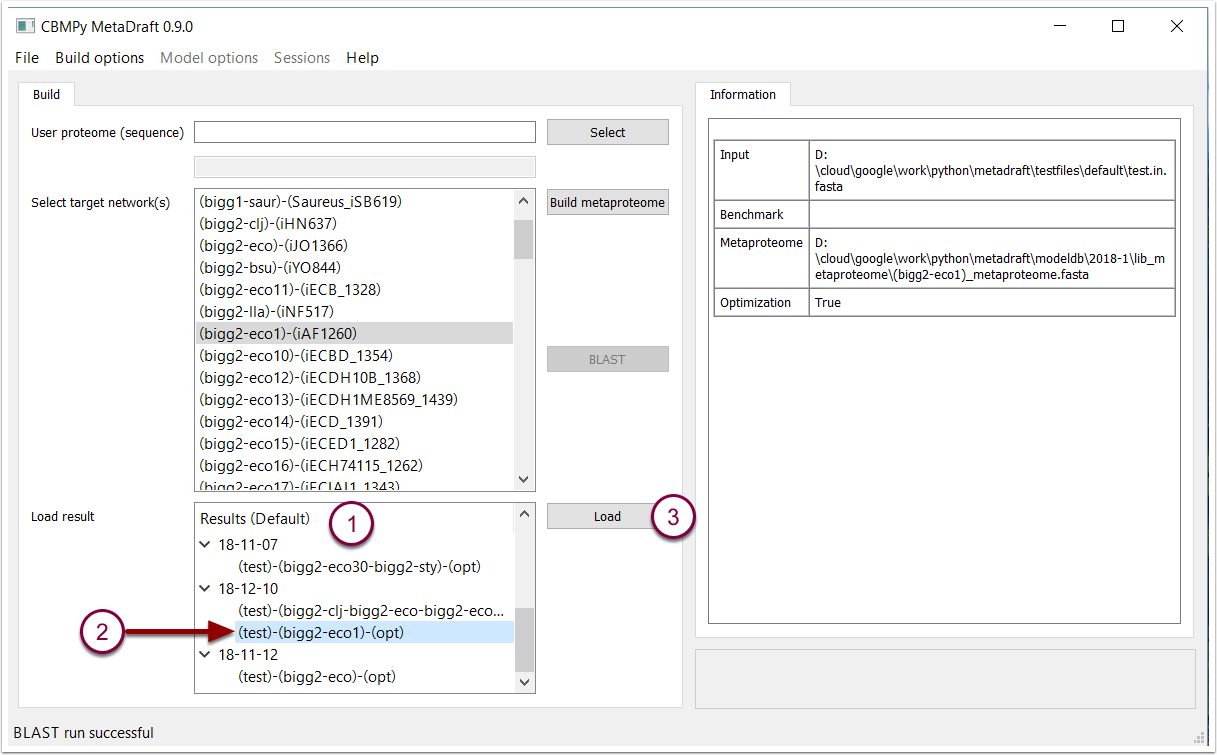
3.1 Delete or rename collections of results
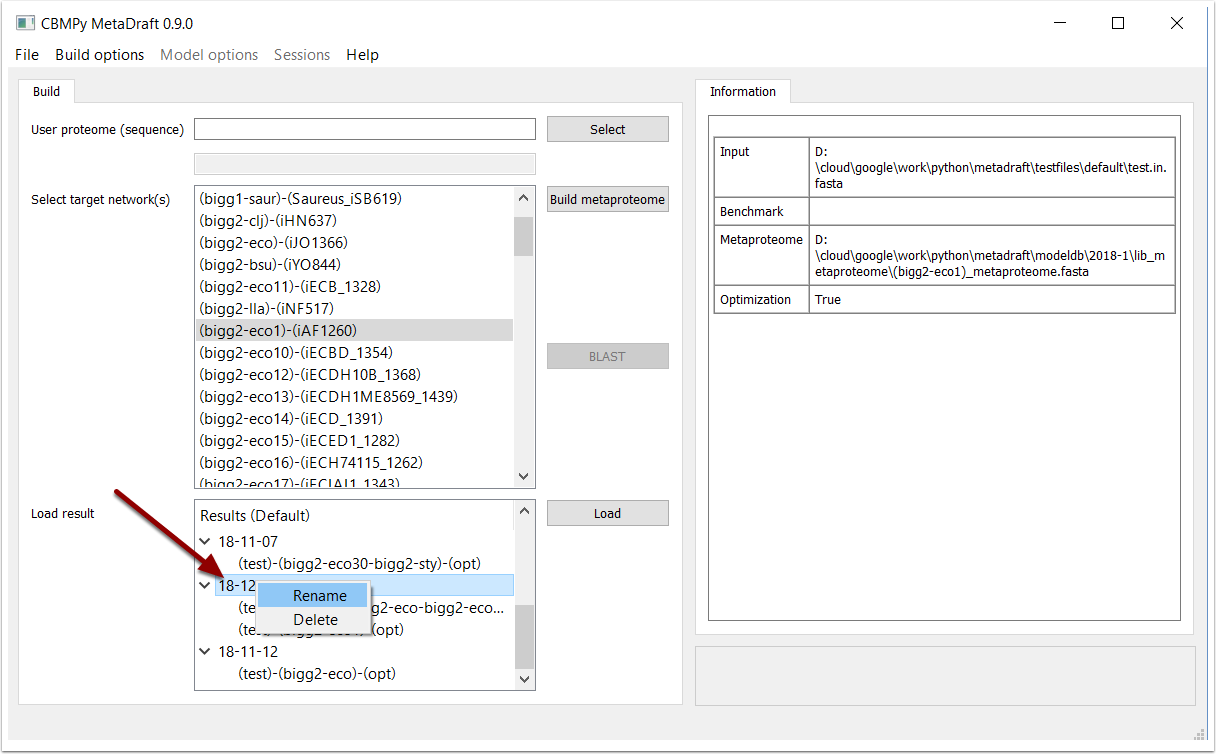
Right-clicking on the result group allows one to either delete the entire group or rename it to somethine more suitable. In addition, right-clicking on a result allows one to delete it, note there is currently no undelete function and deletion is therefore permanent.
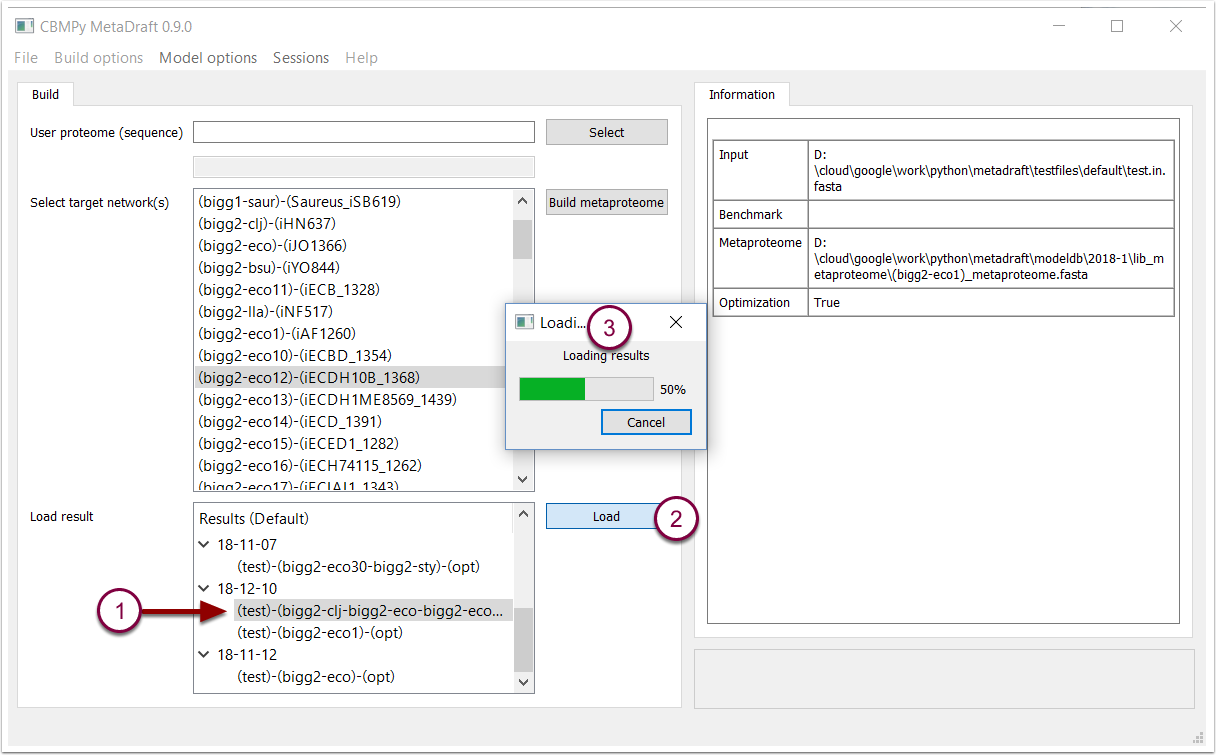
3.2 Loading results for futher reconstruction
Once the results have been generated, they can be loaded for further interpretation and manipulation. This is done by selecting a results set (1), pressing the "Load" button (2) and waiting for the model to load into the edit screen (3).
4. Utility function: analyse and convert an SBML model
MetaDraft template models must be encoded in the latest version of the open, community, standard SBML L3V1 FBCv2. However, many potential template models are encoded in older versions of SBML or SBML dialects, therefore, Metadraft contains functions that assist the modeller in the identification of model type and the creation of template models. To analyse, identify and optionally convert an SBML to the format used by the MetaDraft template generator select "Identify/convert" (1) from the "File" menu.
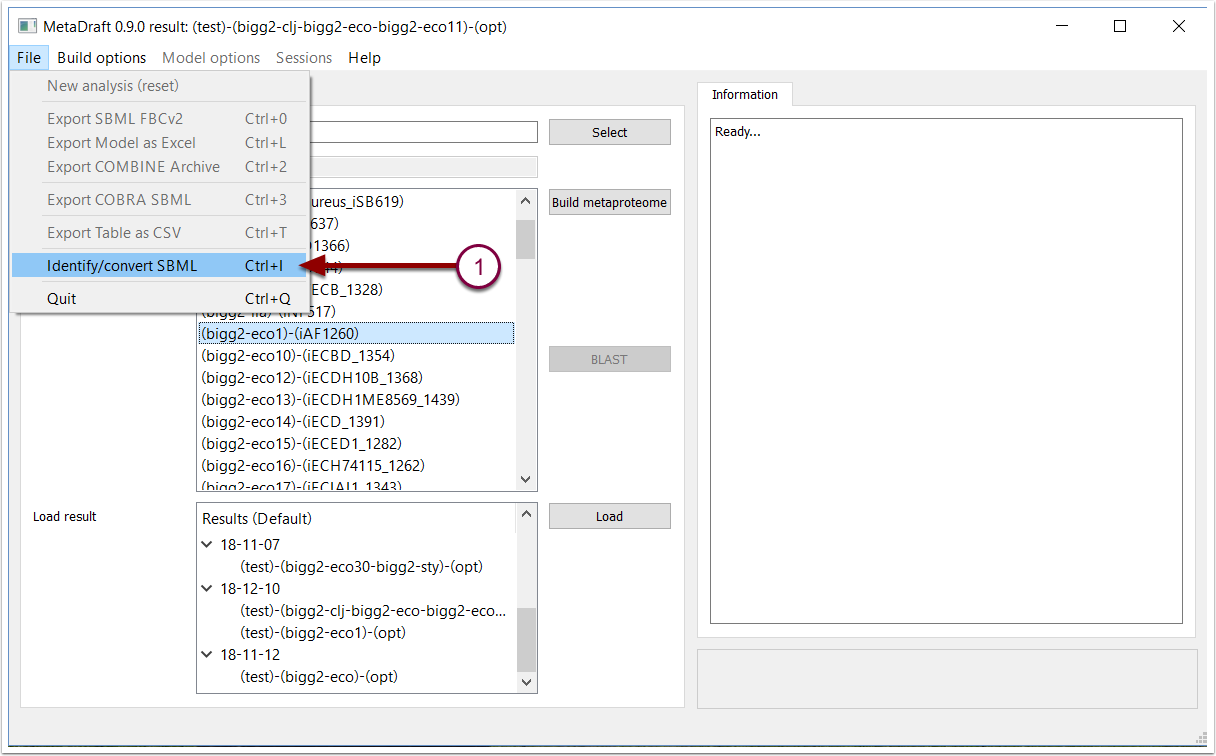
4.1 Utility function: analyse and convert an SBML model
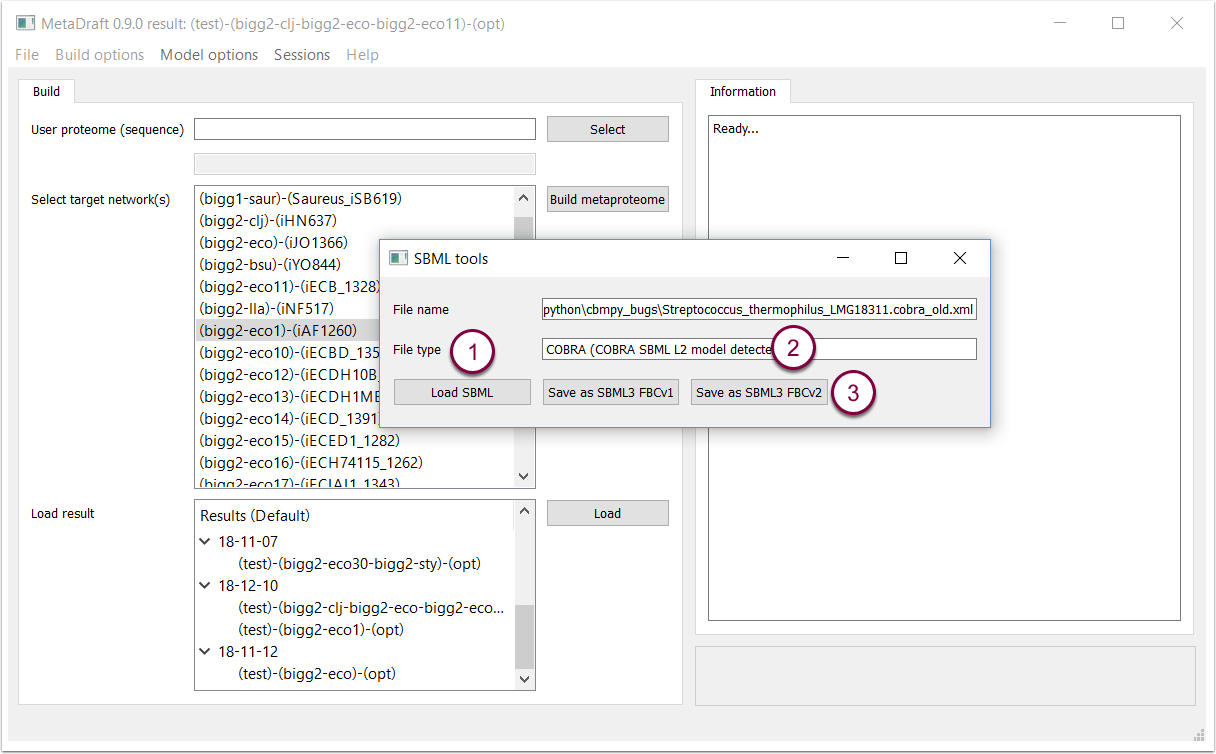
To use the SBML identification/conversion tool, simply load an SBML model (1). If the model encoding is identified this will be displayed in the "File type" field (2) and can then be converted to SBML3 FBCv2 by pressin the "Save as" button (3).
5. Utility function: create a user-defined, template model
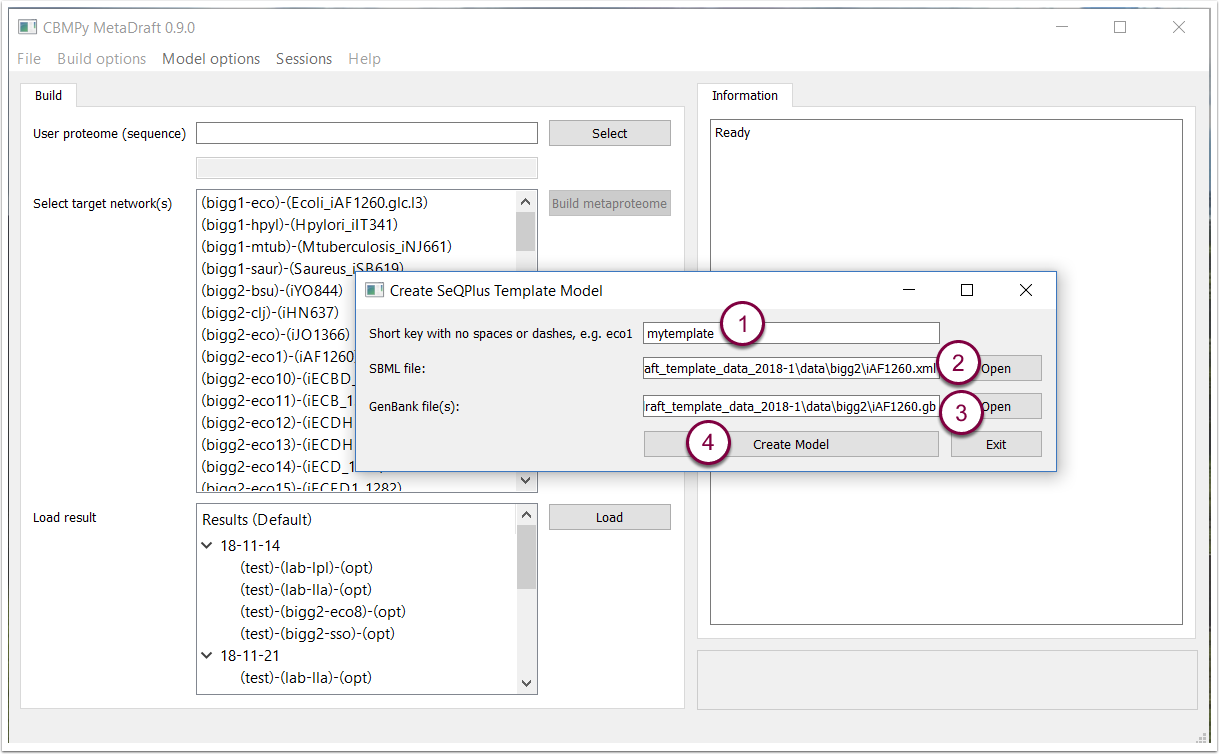
MetaDraft's template database can be extended with the addition of customised SeqPlus templates. These templates are the result of the combination of a genome-scale model (encoded using SBML L3V1 FBCv2 with GPR information) and the associated genome (in GenBank format). Please note that currently, trhe source SBML and GenBank file needs to be located in the same directory. The "Create template model" function is available in the "Build options" menu.
Once selected the template builder window allows you to input a unique model identifier (1), a genome-scale model in SBML format (2) and an associated genbank file that contains CDS annotations.for genes present in the model (3). Once all of the previous information has been entered the template can be created by pressing the "Create Model" button (4). The finals step may take a few minutes.
5.1 Utility function: create a user-defined, template model
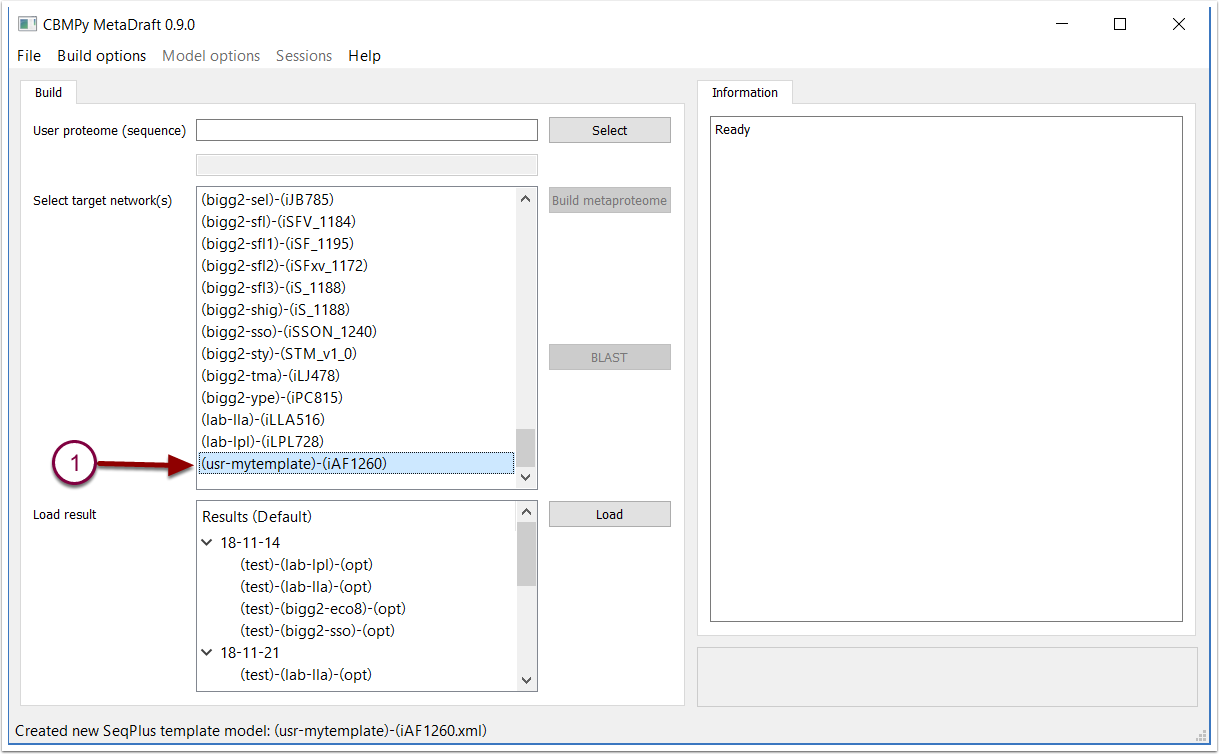
User defined templates are accessible as "(usr-template_id)" in the "Target networks" list.
6. Primary result editing screen: genes
When the results of a BLAST search are loaded they are displayed in MetaDrafts result editing screen. The results are displayed on three interlinked tabs on the left hand side of the panel (1) while any selected components annotation is displayed on the right panel.
Source genes (genes in your input proteome) are displayed in the first column (3), while, where relevant, matches from the metaproteome are displayed in the "match column" (4). The "score" column provides the match score which ranges from 0 to 1 (5) and the "org" column displays the matching template model (6). Finally, the selection column (7) allows genes to be includeded in the final reconstruction or not, for convenience those genes with a 100% match are automatically selected.
Source genes with multiple matches are grouped by colour for easy identification while on the right hand side (2) displays all known annotation about the matching gene and the reactions associated with it. This also includes the GPR association which is colour encoded such that green is a gene that is present and selected, red is present but not selected and black has no match in the target metaproteome (8).
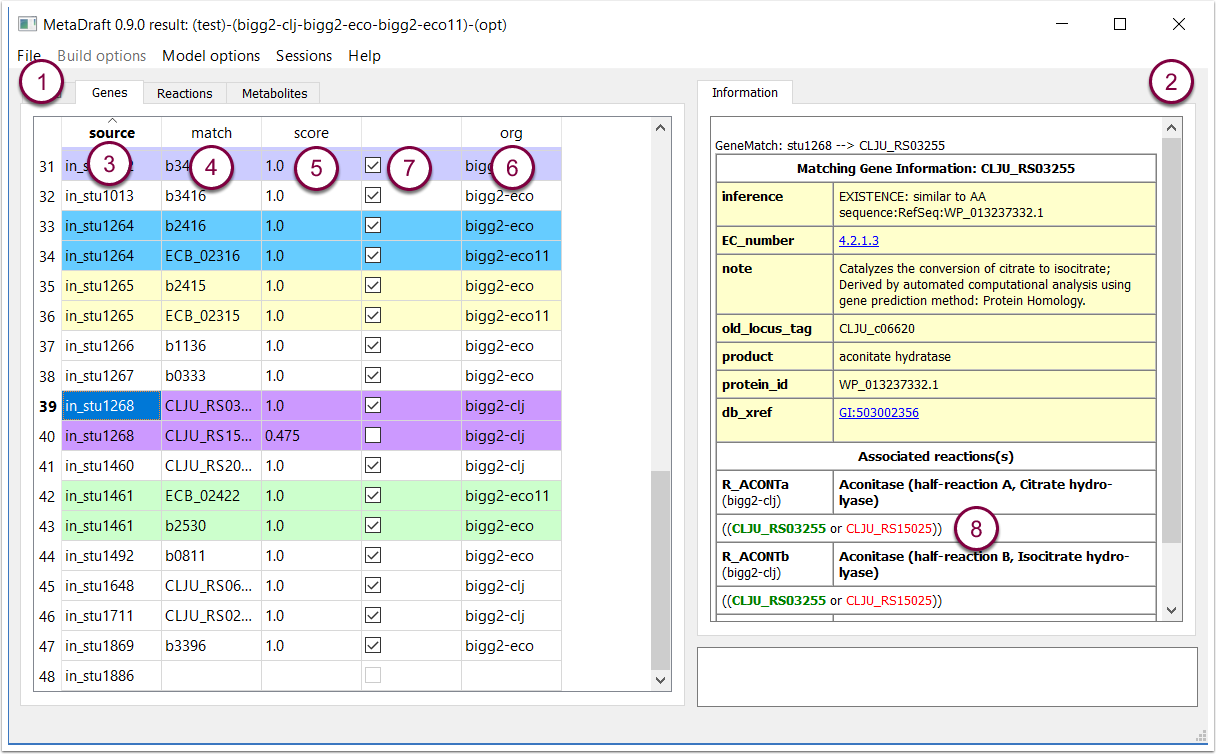
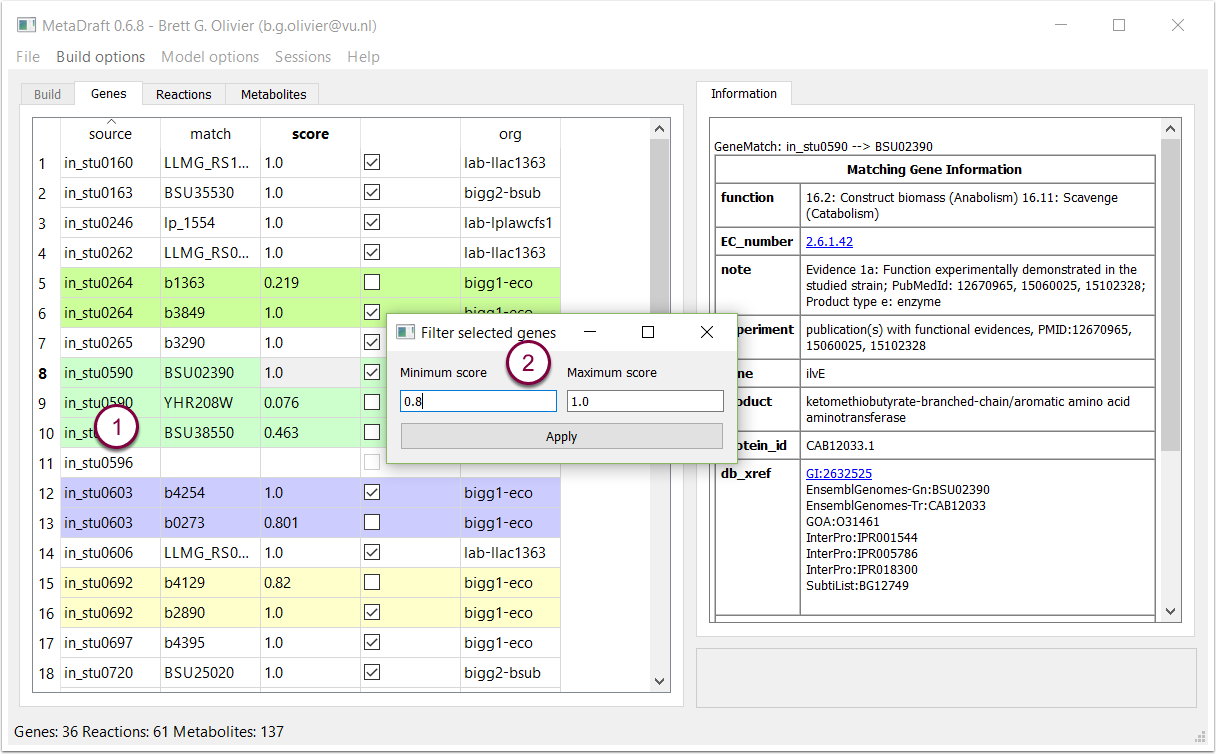
6.1 Gene match score filter
Right clicking on on a row in the "gene" table (1) displays the "Gene Filter" tool where a user defined range matching can be set (between 0 and 1) and applied to quickly select all genes matching the selected score criteria (2).
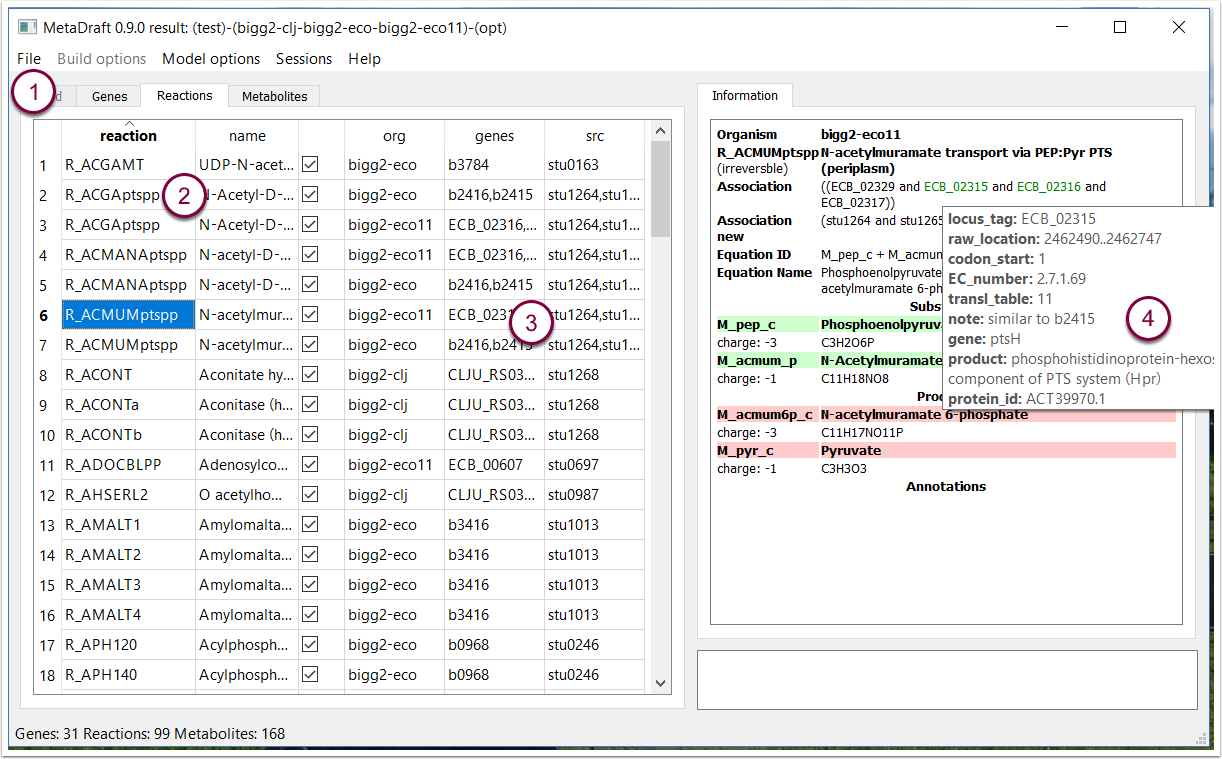
7. Reaction editing screen
As genes are selected in the "Genes table" the corresponding reactions are dynamically displayed in the "Reaction" table (1). Information displayed in the "Reactions" table include the reactions identifier, name and its source model (2). In addition the source gene and its target matches are also provided (3).
On the right hand side any annotation associated with the reaction is displayed including the GPR association given in terms of both the original and newly mapped association (3). Genes are colour encoded to quickly show those that are active, inactive or missing form the original GPR. Hovering over any of the genes in the association displays that genes annotation as a tooltip (4). The model can be further refined by excluding specific reactions form the final reconstruction.
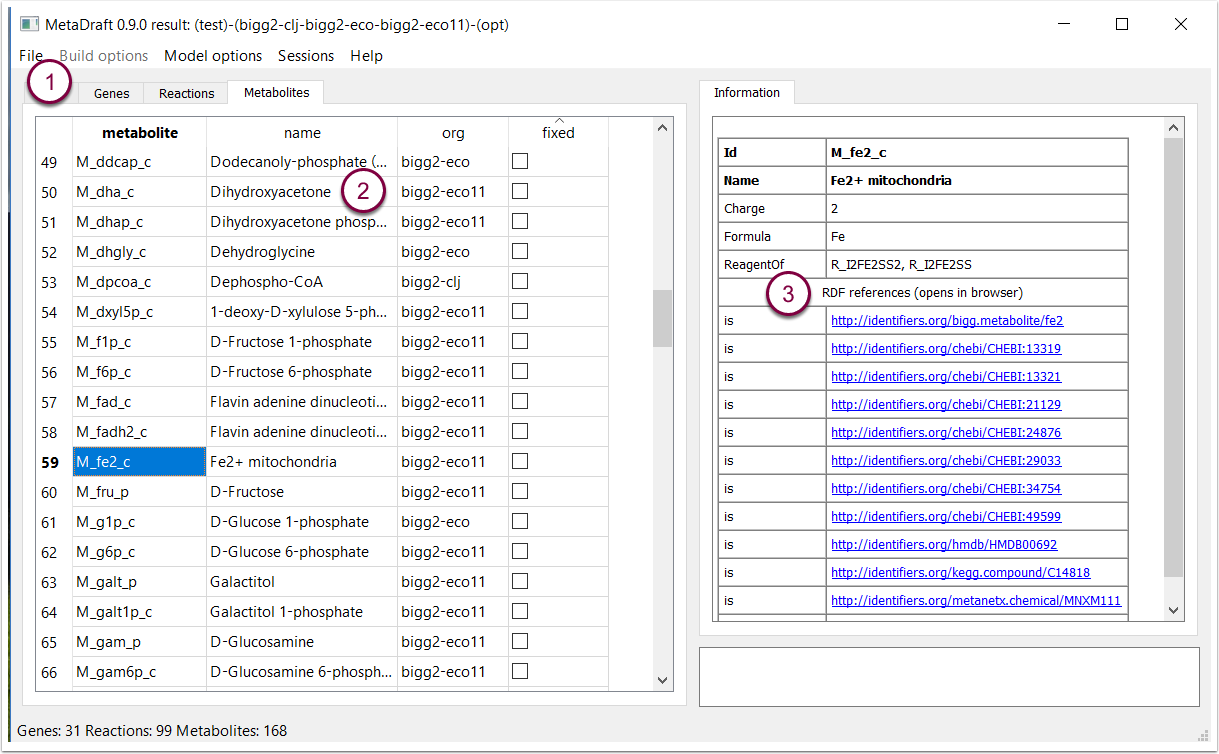
7.1 Metabolite viewer
The metabolite viewer is dynamically updated depending on the reactions selected from the "Reaction" panel (1). The lefthand panel contains metabolite id, name and source model or organism (2). On the right hand side of the viewer any selected metabolite's associated annotation (as defined in the model) is displayed (3). Where possible and if present in the source model, MIRIAM URL's are displayed as live links that can be opened in a webbrowser.
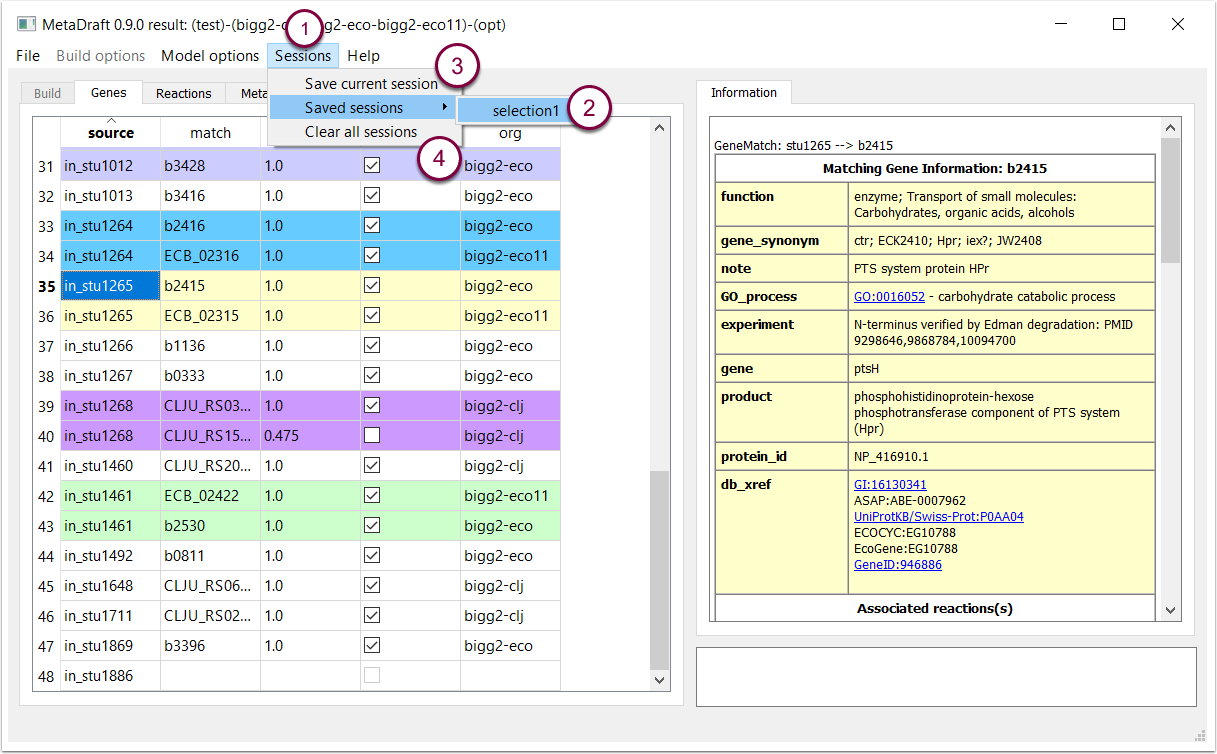
8. Multi-session support
When a result is loaded MetaDraft loads a default selection set which includes the set of selected genes and reactions. Custom selection sets can be created and used in the "Sessions" menu (1). This menu allows you to load (2) and save (3) sessions or clear the list of saved selections (4).
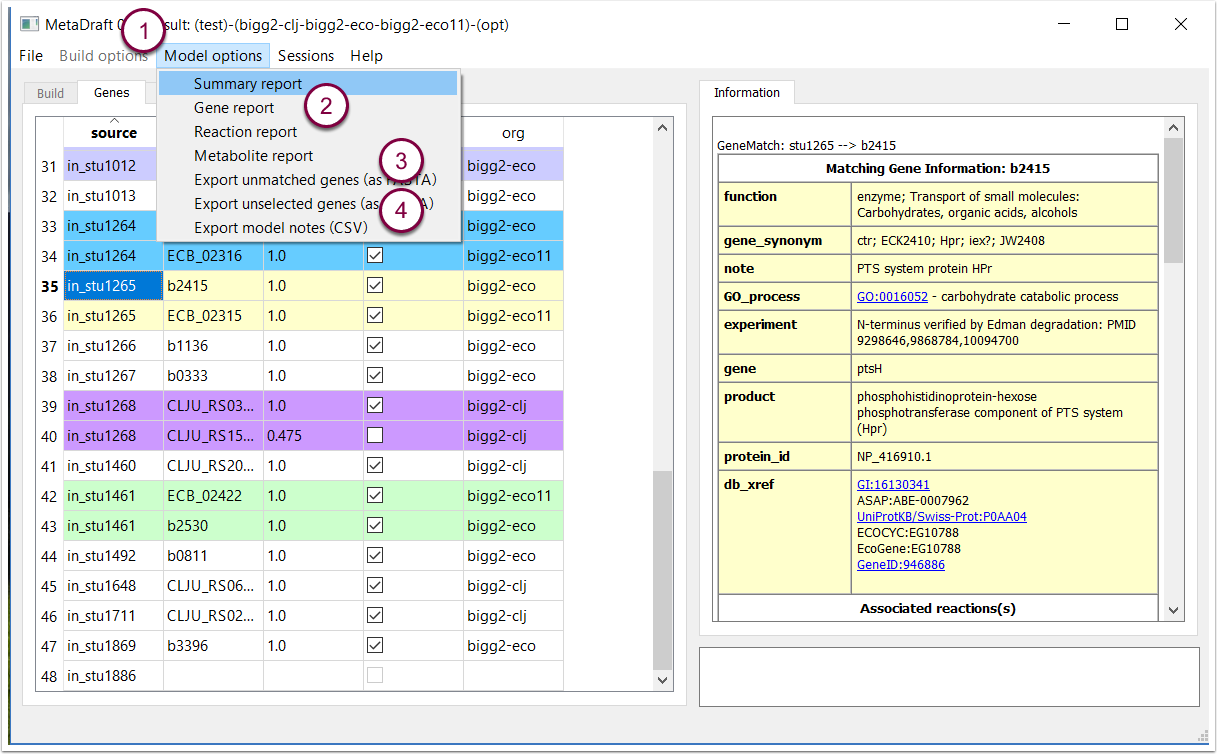
9. Model options and reconstruction reports
Metadraft provides the modeller with an extensive set of reports that detail the contents and components of the draft genome-scale model. These reports are available from the "Model options" menu (1) and include gene, reaction and metabolite reports (2). In addition utility functions are provided that allows the modeller to export (as protein FASTA files) the sets of "unmatched genes" that is genes that do not have any match in the template models (3) as well as unselected genes (4).
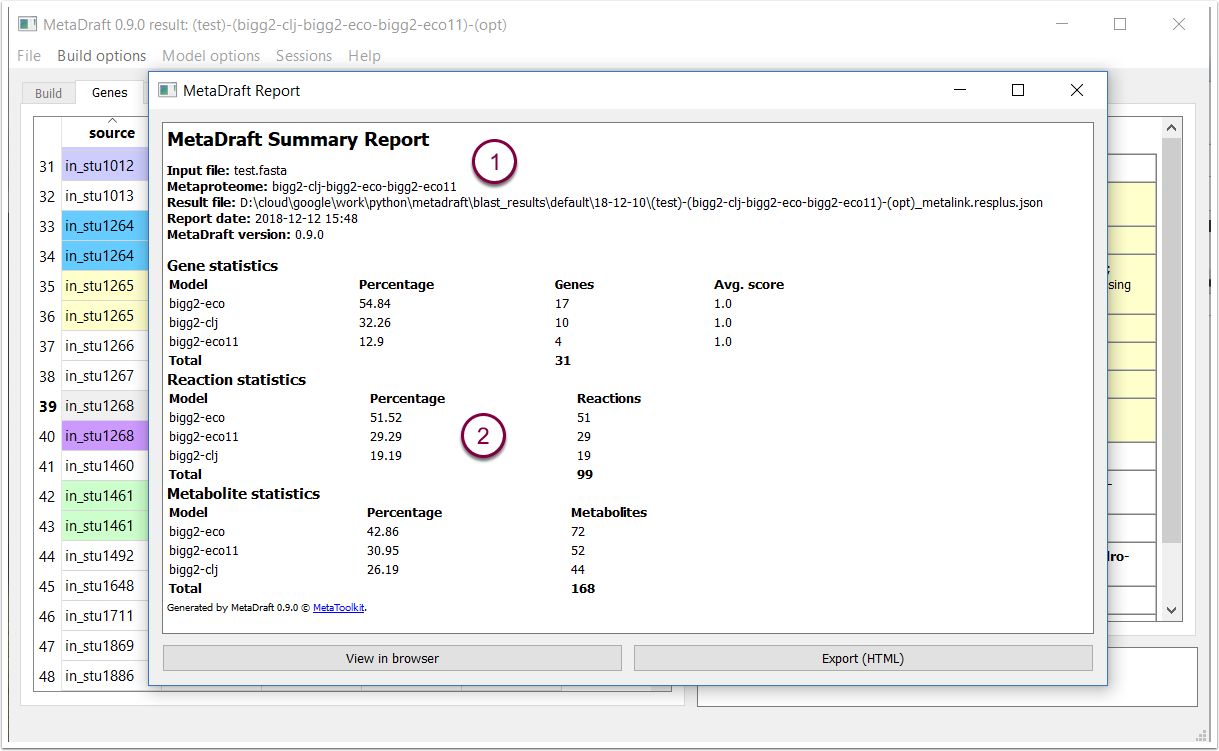
9.1 Report: reconstruction summary
This report shows the source and template models used (1) as well as the overall make up of the new reconstruction. This is provided as a fractional representation of the number of genes, reactions and metabolites, from different sources, that make up the model (2).
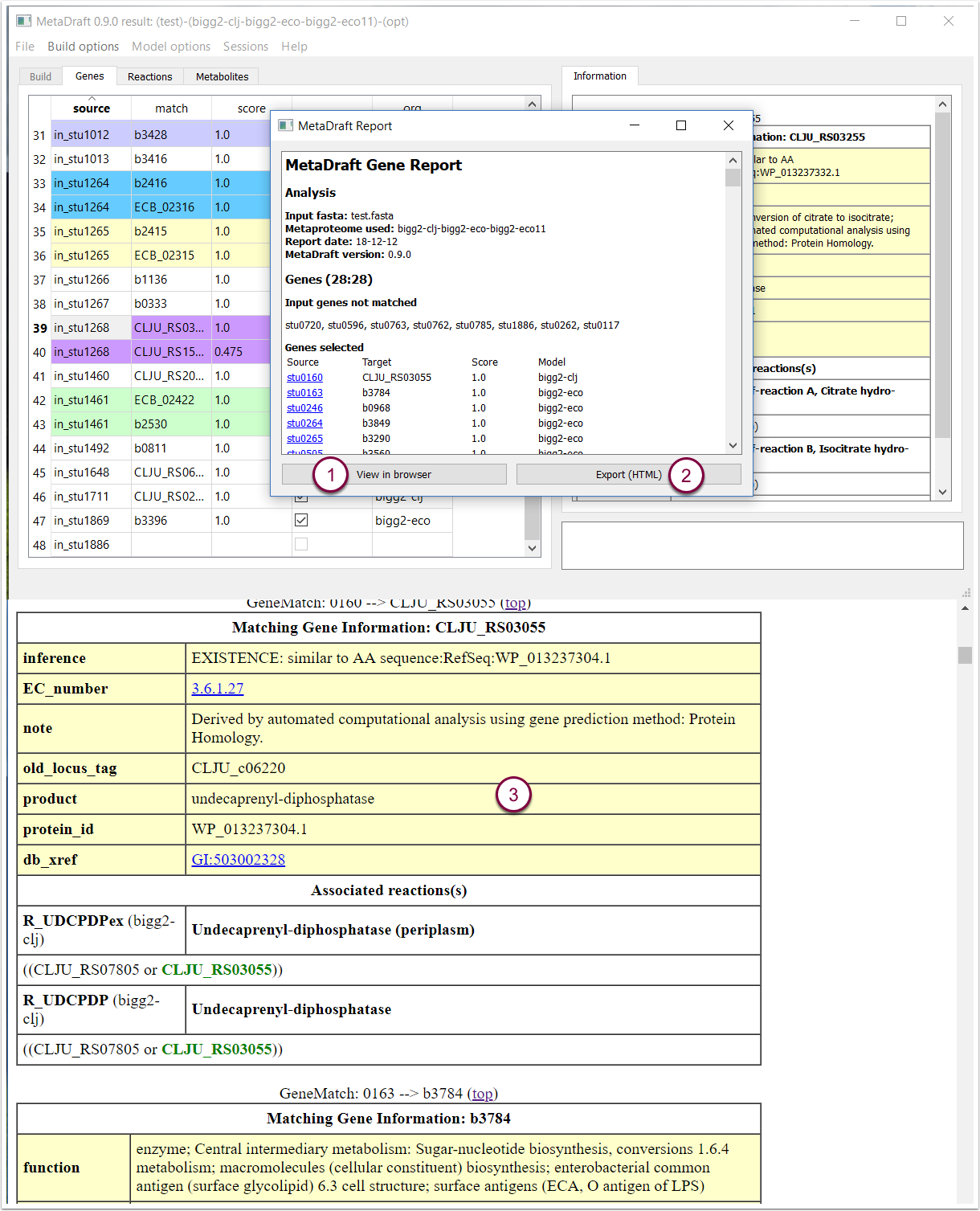
9.2 Report: genes
The "Gene Report" provides details of the input file, metaproteome composition. Source genes with no orthology to the metaproteome are provided together with a table of genes includes in draft reconstruction and their score. In addition the report can be viewd directly in your browser (1) as well as saved as an HTML file (2). The gene id's shown in the report are linked to the gene annotations, show here as they appear in a web browser (3).
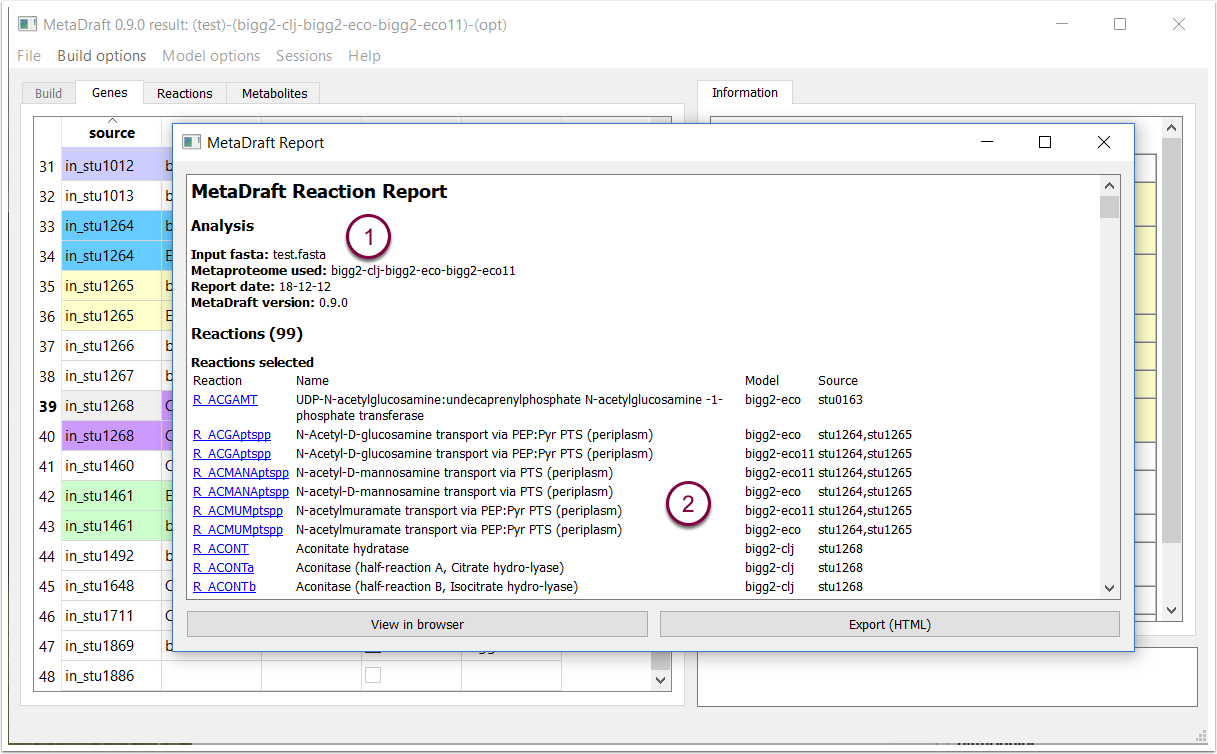
9.3 Report: reactions
The reaction report contains details of the model reconstruction as well as the selected reactions and their annotation.
10. Afterword
Many thanks to all the people that have contributed time in testing, using and providing valuable feedback on earlier versions of MetaDraft.
(C) Systems Bioinformatics, Vrije Universiteit Amsterdam, Amsterdam, 2018.